
Insights



Regulatory–HTA divergence in rare disease outcome assessment: what early evidence planning can do
Rare disease therapies are often developed in settings where trial populations are small, symptoms are heterogeneous, and long-term comparative evidence is difficult to generate. These constraints make outcome selection challenging, particularly when the same evidence package needs to support both regulatory approval and health technology assessment (HTA).
Regulators and HTA bodies may review largely the same clinical evidence but interpret it through different decision-making frameworks. For developers, this can create uncertainty: evidence that is sufficient to demonstrate benefit–risk may not be sufficient to demonstrate added benefit, reimbursement value, or clinically meaningful patient benefit in a specific healthcare system.
This issue is not new, but the introduction of EU Joint Clinical Assessment makes it more time-sensitive.1, 2 As JCA runs alongside regulatory assessment, rare disease developers may need to anticipate HTA questions earlier, before pivotal protocols are finalised and opportunities to generate additional comparative, patient-centred, or long-term evidence become limited.
What we examined
This blogpost expands on our poster presented at the HTAi 2026 Annual Meeting, Regulatory–HTA Divergence in Outcome Assessment for Rare Disease Therapies: Insights from Three European Case Studies. The poster summarised the key divergence themes and presented a condensed framework for early EMA–HTA evidence planning; here, we provide additional context on the case-study findings and the practical implications for rare disease evidence generation.
We explored regulatory–HTA divergence in outcome assessment using three European rare disease case studies previously identified3: solriamfetol4-7 and pitolisant8-10 in narcolepsy, and pegvaliase in phenylketonuria.11-14 The analysis compared how the European Medicines Agency (EMA), Haute Autorité de Santé (HAS), and the Gemeinsamer Bundesausschuss/Institute for Quality and Efficiency in Health Care (G-BA/IQWiG) interpreted clinical evidence, with a focus on outcome-related requirements.
Across the case studies, three recurring areas of divergence were identified:
- comparative context
- outcome interpretation
- evidence credibility
These themes were then used to develop a practical step-by-step framework for early EMA–HTA evidence planning.
Comparative context: efficacy is not the same as comparative value
Comparator expectations were a major source of divergence. EMA may accept placebo-controlled evidence where this is sufficient to establish efficacy and support a positive benefit–risk assessment. In contrast, HTA bodies often require clearer evidence on how a therapy should be positioned against relevant alternatives in routine practice.
Case example: Solriamfetol and pitolisant in narcolepsy
In the narcolepsy case studies, regulatory assessment focused on whether the submitted evidence demonstrated efficacy and an acceptable benefit–risk profile. HTA assessment placed greater emphasis on whether the evidence supported positioning against relevant treatment alternatives in the national reimbursement context. Where direct comparative evidence was limited, uncertainty remained around relative benefit and treatment positioning.
This distinction is particularly important in rare diseases and low-prevalence conditions, where placebo-controlled or single-arm studies may be more feasible than active-comparator trials. However, if the comparator strategy does not reflect expected HTA requirements, the evidence package may later face restrictions, uncertainty, or reduced value recognition in reimbursement decision-making.
Early planning should therefore ask:
- What trial design is likely to be acceptable to EMA?
- What comparator is likely to be considered clinically relevant for HTA or JCA?
- Is a single comparator strategy feasible across regulatory and HTA needs?
- If direct comparative evidence is not feasible, is an indirect comparison, real-world evidence study, or external control arm likely to be feasible?
Where divergence is likely, developers may need to consider active-comparator or multi-arm designs, plan robust indirect comparisons, or prepare clear justification for the selected design.
Outcome interpretation: endpoints need to be meaningful, not just measurable
Outcome interpretation also differed across agencies. EMA accepted disease-specific, functional, and surrogate endpoints where they supported the demonstration of efficacy. HTA bodies more frequently reassessed whether these outcomes were validated, clinically meaningful, patient-relevant, and interpretable in the reimbursement context.
Case example: Pegvaliase in phenylketonuria
EMA accepted reduction in blood phenylalanine as a clinically meaningful indicator of efficacy. HAS and G-BA/IQWiG considered the same endpoint more cautiously, questioning whether biochemical improvement translated into patient-relevant benefit, including neurocognitive outcomes, quality of life, and applicability to routine practice.
This creates a practical challenge for rare disease evidence generation. Surrogate endpoints and disease-specific measures are often necessary because of feasibility constraints, but they require early justification beyond statistical significance or biological plausibility.
Endpoint planning should therefore consider:
- whether each endpoint demonstrates efficacy and supports a patient-relevant benefit;
- whether patient-reported evidence is needed to support interpretation;
- whether surrogate endpoints have a biologically and clinically defensible link to patient benefit;
- whether thresholds for meaningful change, such as minimal clinically important differences or responder definitions, can be defined before submission.
In practice, endpoint strategy should be linked explicitly to patient relevance and HTA interpretability, rather than treated as a purely statistical or regulatory exercise.
Evidence credibility: internal validity is not enough
The third divergence theme related to evidence credibility. HTA bodies placed particular emphasis on whether the trial population, study design, follow-up duration, and supporting evidence were relevant to reimbursement decision-making.
Case example: Pegvaliase in phenylketonuria
EMA accepted evidence from the PRISM trial programme, including responder-enriched and longer-term data, as sufficient to support regulatory conclusions in a rare disease setting. HAS and G-BA/IQWiG were more cautious about the representativeness of the selected trial population and the interpretability of open-label, non-comparative long-term evidence.
For developers, the key issue is not simply whether evidence exists, but whether it is credible for the decision being made. In rare disease settings, trial enrichment, restricted eligibility criteria, small sample sizes, and uncontrolled follow-up may be clinically or operationally unavoidable. However, these features can still limit HTA confidence in generalisability and comparative value.
Early evidence planning should therefore ask:
- Does the study population reflect the reimbursement-relevant population?
- Are eligibility criteria likely to limit generalisability?
- Is follow-up sufficient to demonstrate sustained benefit?
- Will long-term evidence be comparative or primarily uncontrolled?
- What evidence gaps will remain at submission, and how will they be addressed?
These questions are especially important before protocol lock, when there may still be an opportunity to adapt the design, collect additional outcomes, or plan supporting analyses.
A seven-step framework for earlier evidence planning
Based on the divergence patterns identified across the three case studies, we developed a seven-step planning framework to support earlier identification of potential EMA–HTA misalignment.
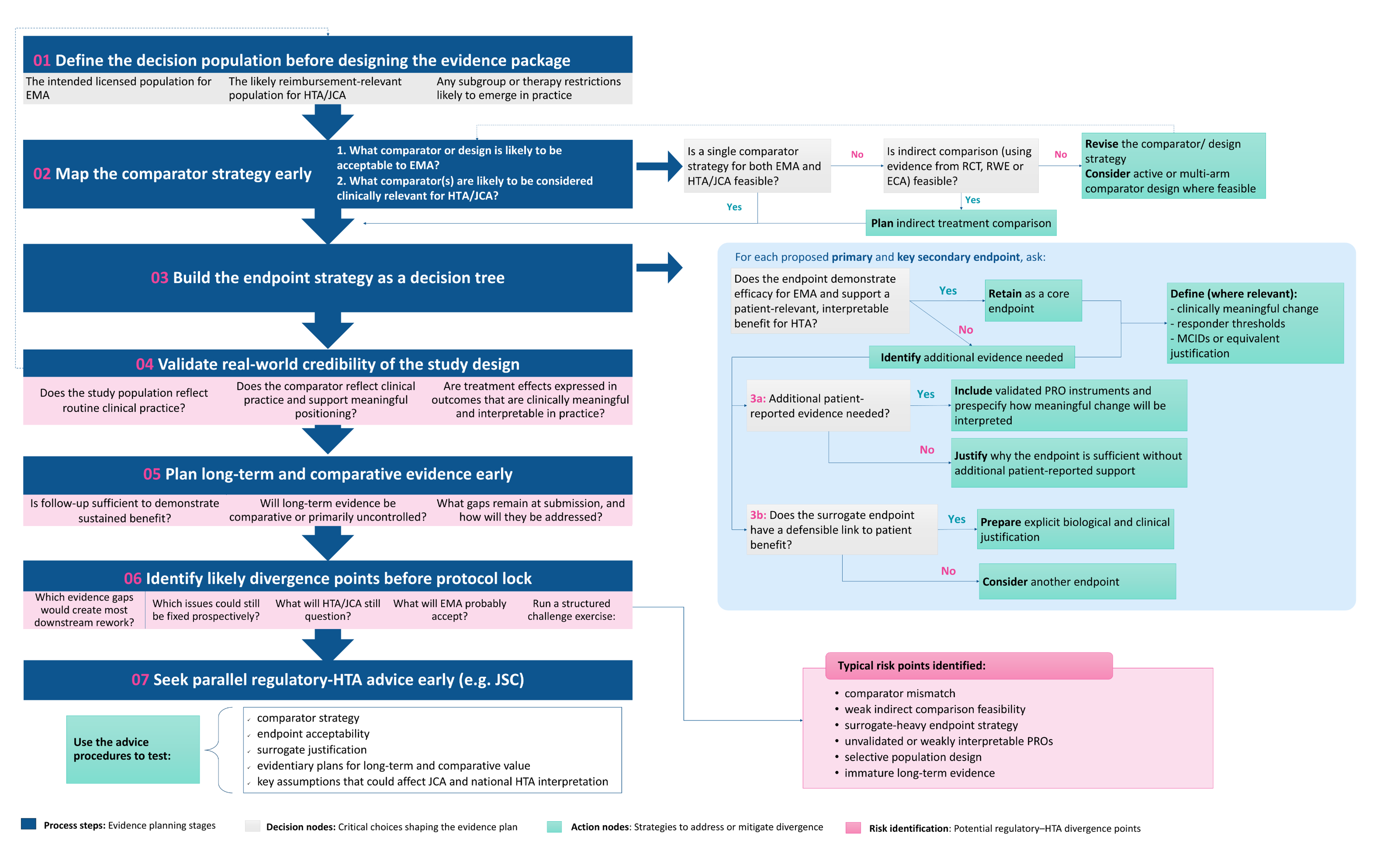
The purpose of the framework is not to suggest that all divergence can be eliminated. In rare diseases, some uncertainty is unavoidable. Rather, the aim is to make divergence more predictable and to identify which evidence gaps can be addressed prospectively.
Preparing rare disease evidence for JCA and HTA
For rare disease therapies, the challenge is not simply to generate more evidence, but to generate evidence that can withstand different regulatory and HTA interpretations. Small patient populations, heterogeneous disease presentation, reliance on surrogate or functional endpoints, limited PRO (patient-reported outcomes) validation, and immature long-term data mean that evidence accepted for regulatory decision-making may still be considered uncertain for reimbursement.
The case studies suggest that a single shared evidence core may be difficult to define under current EMA and HTA frameworks. The same evidence package may support a positive benefit–risk assessment, while still leaving HTA bodies with unresolved questions about the comparator, endpoint relevance, meaningful-change thresholds, and generalisability to routine practice.
This is becoming more important as orphan medicinal products are expected to enter the mandatory JCA scope from January 2028. Evidence planning should consider HTA and reimbursement questions from the start, rather than focusing only on what is needed for regulatory approval. Before pivotal protocols are finalised, developers should stress-test the target population, comparator strategy, endpoint hierarchy, PRO strategy, surrogate justification, follow-up duration, and feasibility of comparative evidence against both EMA and HTA expectations. Joint Scientific Consultation provides a route to test these assumptions earlier by allowing health technology developers to obtain consultation on evidence needs for a subsequent JCA.
Earlier identification of divergence points will not eliminate uncertainty, but it may help avoid predictable evidence gaps, reduce unnecessary rework, and minimise potential delays to patient access.
How can Symmetron help
Symmetron supports developers with early evidence planning for rare disease therapies, including regulatory–HTA evidence strategy, multi-country PICO scoping, endpoint and PRO strategy, evidence-gap assessment, and preparation for Joint Scientific Consultation.
To discuss how early evidence planning could support an upcoming rare disease submission, JCA readiness, or parallel regulatory–HTA engagement, please get in touch with our team.
References
1. European Commission. Joint clinical assessments [Internet]. Brussels: European Commission; [cited 2026 Jun 2]. Available from: https://health.ec.europa.eu/health-technology-assessment/implementation-regulation-health-technology-assessment/joint-clinical-assessments_en.
2. Health Technology Assessment Coordination Group. Guidance on the scoping process for Joint Clinical Assessments [Internet]. Brussels: European Commission; [cited 2026 Jun 2]. Available from: https://health.ec.europa.eu/document/download/7be11d76-9a78-426c-8e32-79d30a115a64_en?filename=hta_jca_scoping-process_en.pdf.
3. Lim V MM, Sawyer L, Langford B,. PCR83 Evaluating the role of disease-specific patient-reported outcomes in HTA submissions for rare conditions: a comparative case study of narcolepsy and phenylketonuria. Value Health. 2025;28.
4. European Medicines Agency. Sunosi: EPAR - medicine overview [Internet]. Amsterdam: European Medicines Agency; [cited 2026 Jun 2]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/sunosi.
5. Haute Autorité de Santé. Sunosi (solriamfetol): transparency committee opinion [Internet]. Saint-Denis: Haute Autorité de Santé; 2020 [cited 2026 Jun 2]. Available from: https://www.has-sante.fr/upload/docs/evamed/CT-18490_SUNOSI_PIC_INS_AvisDef_CT18490_EPI675.pdf.
6. Gemeinsamer Bundesausschuss. Nutzenbewertungsverfahren zum Wirkstoff Solriamfetol (Narkolepsie) [Internet]. Berlin: Gemeinsamer Bundesausschuss; 2020 [cited 2026 Jun 2]. Available from: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/553/.
7. Institute for Quality and Efficiency in Health Care. Solriamfetol (narcolepsy): benefit assessment according to §35a Social Code Book V [Internet]. Cologne: IQWiG; 2020 [cited 2026 Jun 2]. Available from: https://www.iqwig.de/en/projects/a20-47.html.
8. European Medicines Agency. Wakix: EPAR - medicine overview [Internet]. Amsterdam: European Medicines Agency; [cited 2026 Jun 2]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/wakix.
9. Haute Autorité de Santé. Wakix (pitolisant): narcolepsy [Internet]. Saint-Denis: Haute Autorité de Santé; [cited 2026 Jun 2]. Available from: https://www.has-sante.fr/jcms/p_3603851/en/wakix-pitolisant-narcolepsy.
10. Gemeinsamer Bundesausschuss. Pitolisant: resolution on the amendment of the Pharmaceuticals Directive, Annex XII [Internet]. Berlin: Gemeinsamer Bundesausschuss; 2023 [cited 2026 Jun 2]. Available from: https://www.g-ba.de/downloads/91-1455-938/2023-09-21_Current-Version_Pitolisant_D-916_EN.pdf.
11. European Medicines Agency. Palynziq: EPAR - medicine overview [Internet]. Amsterdam: European Medicines Agency; [cited 2026 Jun 2]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/palynziq.
12. Haute Autorité de Santé. Palynziq (pegvaliase): phenylketonuria [Internet]. Saint-Denis: Haute Autorité de Santé; [cited 2026 Jun 2]. Available from: https://www.has-sante.fr/jcms/p_3221966/en/palynziq-pegvaliase.
13. Gemeinsamer Bundesausschuss. Nutzenbewertungsverfahren zum Wirkstoff Pegvaliase (Phenylketonurie) [Internet]. Berlin: Gemeinsamer Bundesausschuss; 2019 [cited 2026 Jun 2]. Available from: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/471/.
14. Institute for Quality and Efficiency in Health Care. Pegvaliase (phenylketonuria): assessment according to §35a Social Code Book V [Internet]. Cologne: IQWiG; 2019 [cited 2026 Jun 2]. Available from: https://www.iqwig.de/projekte/g19-12.html.
Similar Insights





.jpg)

















Stay in touch
Subscribe to Symmetron and stay up to date with recent news and announcements.


8 Devonshire Square,
London, EC2M 4PL, UK




